Integrating virus genomics into public health
Virus evolution and diversity can impact outbreak dynamics and control efforts. This has been thoroughly demonstrated during COVID-19 pandemic, in particular with the emergence of variants. Monitoring evolutionary processes can provide answers that are hidden from traditional epidemiology, both on a small scale by investigating the details of transmission chains and uncovering the pathways of spread on a global scale. Genomic data also has the potential to enhance disease forecasting models and has been critical for development of vaccines, therapeutics, and molecular diagnostic assays. The overall goal of our lab is to apply virus sequencing and genomic analysis to better design, apply, and evaluate strategies to mitigate transmission and disease. While our primary interests are mosquito- and tick-borne viruses, like dengue, West Nile, and Powassan, we are broadly interested in genomic epidemiology across many viruses, including SARS-CoV-2.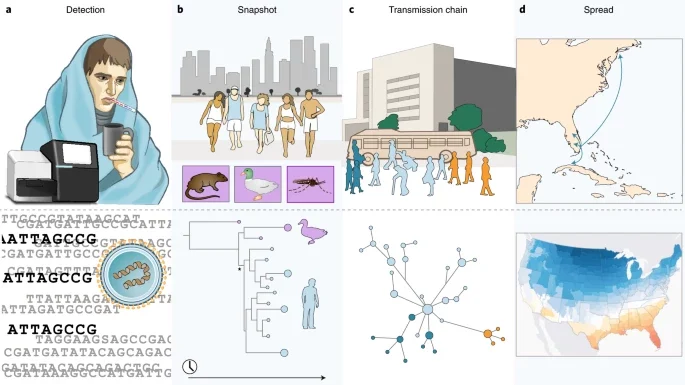
Tracking outbreaks using genomics
Genomics has become an important tool to track the emergence and spread of viruses (Grubaugh et al., 2019). At the core of our lab is the routine sequencing of viruses to facilitate public health decision making and epidemiological investigations. When SARS-CoV-2 emerged in the US, we partnered with the Yale New Haven Hospital and the Connecticut Department of Public health to create the Yale SARS-CoV-2 Genomic Surveillance Initiative (CovidTrackerCT). Our data provided real-time information to the public about the emergence of variants and important data to understand the epidemiology of the virus. For example, sequencing the first COVID-19 cases from the state revealed that the introductions were likely from domestic rather than international locations (Fauver et al., 2020), and expanded sequencing revealed the patterns of introductions of new variants (Alpert et al., 2021). We also built a framework to connect our sequencing data to other epidemiological data to measure the transmissibility of variants (Petrone et al., 2022; Earnest et al., 2022). Looking forward, our group received a prestigious NIH New Innovator Award in 2022 to apply these techniques to better understand the dynamics of dengue virus. Following the Zika epidemic, we discovered that the dengue resurgence was caused by cryptic viruses circulating locally for years (Brito et al., 2021). With more routine sequencing, we aim to discover how the emergence of dengue virus lineages is associated with outbreaks. To do these projects, we will collaborate with many awesome labs from around the world (see a list of our collaborators at the bottom of the “team” page).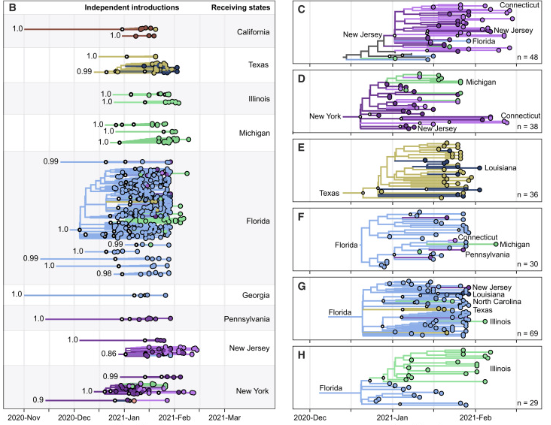

Impact of genetic diversity on control measures
Virus evolution and genetic diversity can significantly impact control measures, as clearly demonstrated during the COVID-19 pandemic. In particular, we showed how new variants can alter serum neutralization from vaccinated individuals (Lucas et al., 2021) and have higher potential to cause vaccine breakthrough infections (Chaguza et al., 2022). We are now working with colleagues at the Yale School of Medicine and Public Health to routinely track the impact of SARS-CoV-2 variants on vaccines and outbreak potential. As vaccines and other control measures are developed and deployed for dengue and other viruses, we must adapt these systems to study the impact of genetic diversity. For example, we are using both experimental and field-based designs to determine how dengue virus diversity impacts the transmission control of Wolbachia-infected mosquitoes, and we aim to apply the same framework to study dengue vaccine candidates.Ecological drivers of emergence and spread
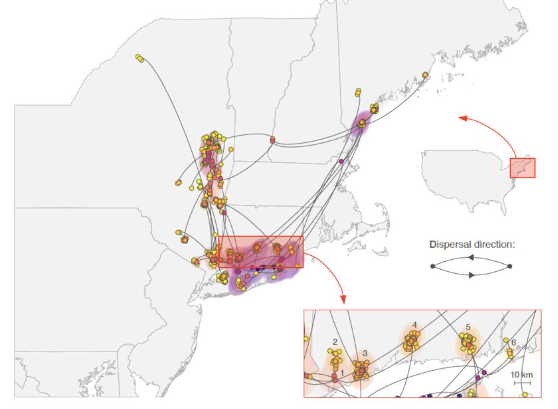
Mosquito and tick-borne viruses have complex ecologies which can define the speed and range of their spread. Several years ago, we started the WestNile4K project to sequence West Nile virus from throughout the Americas to better define “transmission zones” and the ecological barriers that define them. Our work revealed the initial rapid spread of West Nile virus across the US and how it evolved into distinct lineages (Hadfield et al., 2019). Using this network, we expanded to include eastern equine encephalitis virus and the tick-borne Powassan virus. Our sequencing and phylogenetics revealed the (re)emergence of Powassan virus into the Northeast US and that transmission is limited to tight geographical foci (Vogels et al., 2022). Furthermore, we are incorporating environmental data into mosquito-borne virus transmission models to better inform outbreak predictions.